
sD Distrofias Musculares da Cintura
Diisferlinopatia
L. Negrão1, A. Geraldo1, O. Rebelo1, A. Marques1, R. Santos2
Sinapse 2005;5:89.
1 Consulta Externa de Doenças Neuromusculares do Serviço de Neurologia, HUC, Coimbra; 2 Unidade de Genética Molecular do Instituto de Genética Médica Jacinto Magalhães, Porto
Introdução: A disferlina é uma proteína da membrana plasmática muscular e o gene da disferlina localiza-se no cromossoma 2p13. Três fenótipos distintos estão descritos secundários à sua ausência ou expressão reduzida, nomeadamente a Distrofia Muscular das Cinturas 2B (DMC 2B), a Miopatia de Miyoshi (MM) e a Miopatia Distal do Compartimento Anterior (MDCA).
Objectivos: Apresentar as características clínicas, laboratoriais e evolução clínica de oito doentes com doença muscular devida a uma deficiência da proteína disferlina.
Material e Métodos: Análise dos processos clínicos de oito doentes com sinais e/ou sintomas clínicos secundários a um defeito da proteína disferlina, confirmado por técnicas de imunohistoquímica e/ou genética molecular.
Resultados: Os oito doentes dividem-se em número igual por ambos os sexos. O tempo médio de evolução desde os primeiros sintomas até à actualidade é de 10,2 ± 6,5 anos (2-24 anos) e a idade média actual é de 29,5 ± 9,1 anos (20-49 anos). Na observação clínica inicial, um doente apresentava sintomas de fatigabilidade muscular (EF), o fenótipo DMC 2B observava-se em cinco doentes e o fenótipo MM em dois doentes. Actualmente, quatro doentes apresentam agravamento com generalização da fraqueza muscular (3 DMC 2B e 1 MM) e os restantes mantêm a gravidade do quadro clínico inicial (2 DMC 2B, 1 MM e 1 EF).
Os valores séricos de CK estavam elevados em todos os doentes, identificou-se um padrão do tipo distrófico em sete biopsias musculares, o estudo com anticorpo anti-disferlina foi positivo nos cinco doentes em que foi utilizado e mutações patogénicas no gene da disferlina confirmaram-se em sete doentes, observando-se a mesma mutação em três doentes, dois com o fenótipo DMC 2B e um com o fenótipo MM.
Conclusão: O conjunto de doentes apresentados demonstra a reconhecida heterogeneidade, clínica e genética, das doenças musculares associadas a um defeito da disferlina, salientando-se também a variabilidade da gravidade e evolução clínicas.
Palavras-chave: Disferlinopatia; Disferlina; Miopatia de Miyoshi; Distrofia Muscular das Cinturas
Artigo completo: aqui.
Distrofia Muscular das Cinturas 2B
No sistema de classificação ternário das doenças musculares proposto por Walton e Nattrass em 1954 (1), as Distrofias Musculares das Cinturas (DMC) eram definidas como doenças hereditárias do músculo caracterizadas por fraqueza e atrofia muscular progressiva e que apresentavam, na histologia muscular, necrose e degeneração da fibra muscular e invasão por tecido conectivo e gordura. Nesta classificação, os autores pretendiam individualizar as “novas” doenças musculares hereditárias de duas outras doenças bem caracterizadas clínica e histologicamente no século XIX, a Distrofia Facioescapulohumeral e a Distrofia Muscular de Duchenne.
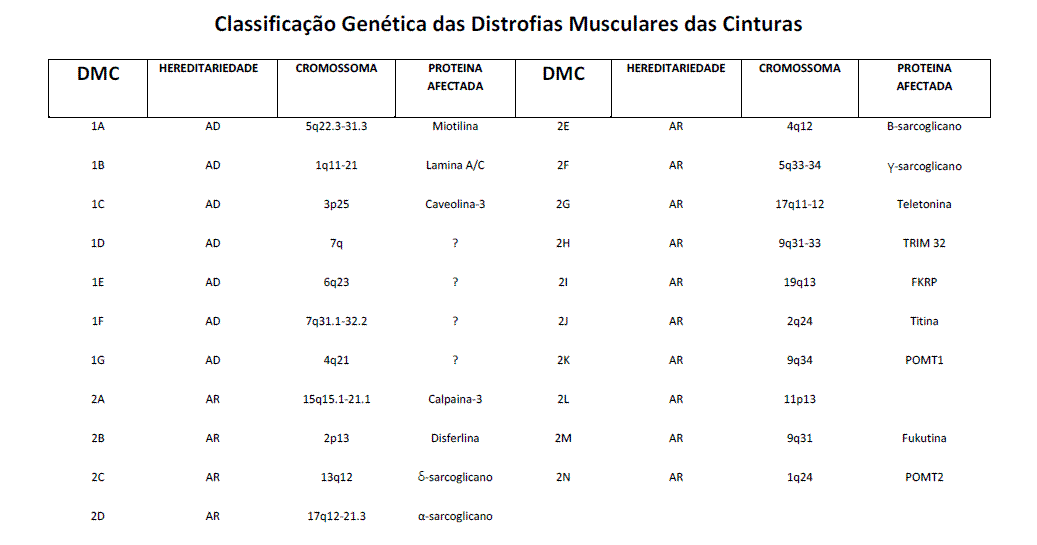
O Consórcio sobre DMC do Centro Europeu de Doenças Neuromusculares (ENMC), reunido em 1995 (2), actualizou o conceito e estabeleceu os critérios de diagnóstico actuais. As DMC são definidas como doenças musculares hereditárias progressivas, com predomínio proximal da fraqueza muscular (cinturas escapular e pélvica), preservando a face e associando elevação da enzima muscular creatina kinase (CK) e aspectos distróficos na biopsia muscular. Foram classificadas em dois grandes grupos, consoante o modo de transmissão, autossómica dominante (AD) ou autossómica recessiva (AR); o primeiro foi designado de 1 e o segundo de 2, apresentando cada um dos grupos vários subtipos, designados por uma letra de acordo com a ordem cronológica da descoberta do gene (DMC 1A, 1B, 1C, 2A, 2B, etc.). As formas AR são as mais comuns, compreendendo 90% das DMC. Catorze genes nas formas AR e três nas formas AD foram clonados e os produtos proteicos identificados. Em quatro formas AD, o locus cromossómico foi localizado mas não foi identificado nem o gene nem o produto proteico. Os genes responsáveis por estas proteínas distribuem-se por diferentes cromossomas e em diferentes áreas de um cromossoma.
O diagnóstico das DMC é cada vez mais complexo e difícil. Algumas das razões que explicam esta dificuldade são:
1- Uma mutação patogénica no gene responsável por uma determinada proteína da fibra muscular pode apresentar manifestações clínicas diferentes (heterogeneidade clínica). Esta situação acontece particularmente nas mutações dos genes da disferlina, calpaina-3 e caveolina-3.
2- Noutros casos, a mesma manifestação clínica pode ser o resultado de mutações patogénicas em genes diferentes (heterogeneidade genética). 3- As várias proteínas responsáveis pelas diferentes formas de DMC são constituintes da fibra muscular e localizam-se em diferentes áreas da fibra muscular. No sarcolema (distrofina, disferlina, sarcoglicanos e caveolina-3), envelope nuclear (lamina A/C), matrix extracelular (laminina 2, colagénio VI), sarcómero (teletonina, titina, miotilina) e sarcoplasma (calpaina- 3, TRIM32¸ -distroglicanos).
4- A variabilidade na gravidade e na forma de apresentação clínica dos diferentes subtipos:
a) as primeiras manifestações clínicas das DMC podem surgir na infância (laminina na adolescência (disferlina) ou na vida adulta;
b) a evolução clínica pode ser rápida, com perda da capacidade de marcha autónoma ou lentamente progressiva;
c) podem complicar-se de insuficiência cardíaca e/ou respiratória, que pode ser frequente e grave (sarcoglicanos e -distroglicanos) ou nunca ocorrer (disferlina);
d) a contractura articular, nalguns casos generalizada e incapacitante, pode ser a primeira manifestação da doença (lamina A/C) ou surgir nas fases avançadas da doença, resultado da imobilidade articular por fraqueza muscular.
e) algumas formas de DMC apresentam em simultâneo com a atrofia muscular proximal, hipertrofia muscular focal significativa (sarcoglicanos, -distroglicanos);
f) outras doenças apresentam a fraqueza muscular inicial predominando nos músculos distais dos membros inferiores (disferlina e titina).
O diagnóstico das DMC é baseado na história clínica, no exame neurológico, no estudo laboratorial (CK), na avaliação cardio-respiratória, na biopsia muscular e no estudo de genética molecular. A biopsia muscular é fundamental na avaliação diagnóstica, permitindo avaliar directamente as características histológicas gerais típicas das DMC e, através das técnicas de imunohistoquímica e quantificação da proteína em estudo (“imunoblot”), confirmar a ausência da proteína muscular em estudo e quantificar essa proteína nas situações em que ela é evidenciada mas aparentemente em quantidade reduzida, respectivamente. O estudo de genética molecular, orientado pelos resultados dos testes anteriores, é o teste diagnóstico definitivo ao revelar uma mutação patogénica no gene codificante da proteína em estudo e, assim, confirmando a doença inicialmente suspeitada.
Actualmente, nenhuma das DMC tem cura. As medidas terapêuticas disponíveis do tipo conservador visam diminuir as consequências da fraqueza muscular na qualidade de vida do doente neuromuscular e prolongar a sobrevida. Ajudas técnicas para melhorar a autonomia motora, a correcção cirúrgica de deformidades esqueléticas (escoliose por ex.), o controlo das manifestações de insuficiência cardio-respiratória e o tratamento da dor são alguns exemplos das terapêuticas disponíveis e que devem ser individualizadas para cada doente. Nos últimos anos tem sido desenvolvida uma intensa investigação científica pluridisciplinar e multicêntrica com o objectivo de curar as DMC. Com a aplicação de sofisticadas técnicas de engenharia genética pretende-se restaurar a normal expressão e função da proteína no músculo e, desta forma, restaurar a função motora perdida. No caso das situações de diagnóstico préclinico, as terapêuticas genéticas podem impedir o aparecimento da sintomatologia clínica.
É provável que se assista na segunda década deste século a uma revolução no tratamento das doenças geneticamente determinadas e, em particular, das DMC.
Bibliografia:
1- Walton, JN, Nattrass FJ. On the classification, natural history and treatment of the
myopathies. Brain 1954;77:169-231.
2- Bushby KMD, Beckmann JS. The limb girdle muscular dystrophies-Proposal for a new
nomenclature. Neuromuscul Disord 1995;5:337-343.
Classificação Genética das Distrofias Musculares das Cinturas
Luis Negrão, Assistente Hospitalar Graduado de Neurofisiologia
Coordenador do Laboratório de Electromiografia e Potenciais Evocados e da Consulta Externa de Doença Neuromusculares do Serviço de Neurologia dos Hospitais da Universidade de Coimbra.
P.S. Texto elaborado para o Manual do Cuidador do Doente Neuromuscular